Data Analysis Tools
The two primary analysis performed at the UWPR are shotgun or bottom up proteomics and targeted quantitative proteomics.
For targeted proteomics, data is classically acquired on triple quadrupole instruments and typically analyzed using the Skyline software suite. There are extensive documentation, tutorials, and videos on the Skyline web site. The nice thing about Skyline is the develop is done here at the UW in the MacCoss lab in Genome Sciences (in the Foege building on South campus).
Shotgun analysis involves peptide identification via MS/MS database searching. Popular commercial tools are Thermo's Protein Discoverer and Proteome Software's Scaffold. Free tools such as MaxQuant and FragPipe are widely used for performing peptide ID, quantification, and more.
To use the same tools as we do here at the UWPR, you can learn about the Trans-Proteomic Pipeline (TPP) originally developed in the Aebersold group at ISB. There's a support forum for your questions and they offer periodic week-long software courses if you want to learn how to use the software.
Data analysis primer
Shotgun data processed by the UWPR for you will typically entail a Comet database search followed by Trans-Proteomics Pipeline (TPP) analysis using PeptideProphet and ProteinProphet. Aimed at someone new to UWPR proteomics analysis, this is a brief tutorial on how to start looking at your data.
Links to your processed data will appear at the bottom of your project page in the section "External Links to Data".
Click on the "View Data" link which will bring up a page that looks like this:
Clicking on these links will prompt you to authenticate again using the same user name and password that gets you into your UWPR project page. If you're sure you're using the same login credentials but are still not able to access the data links, please try the instructions on this page.
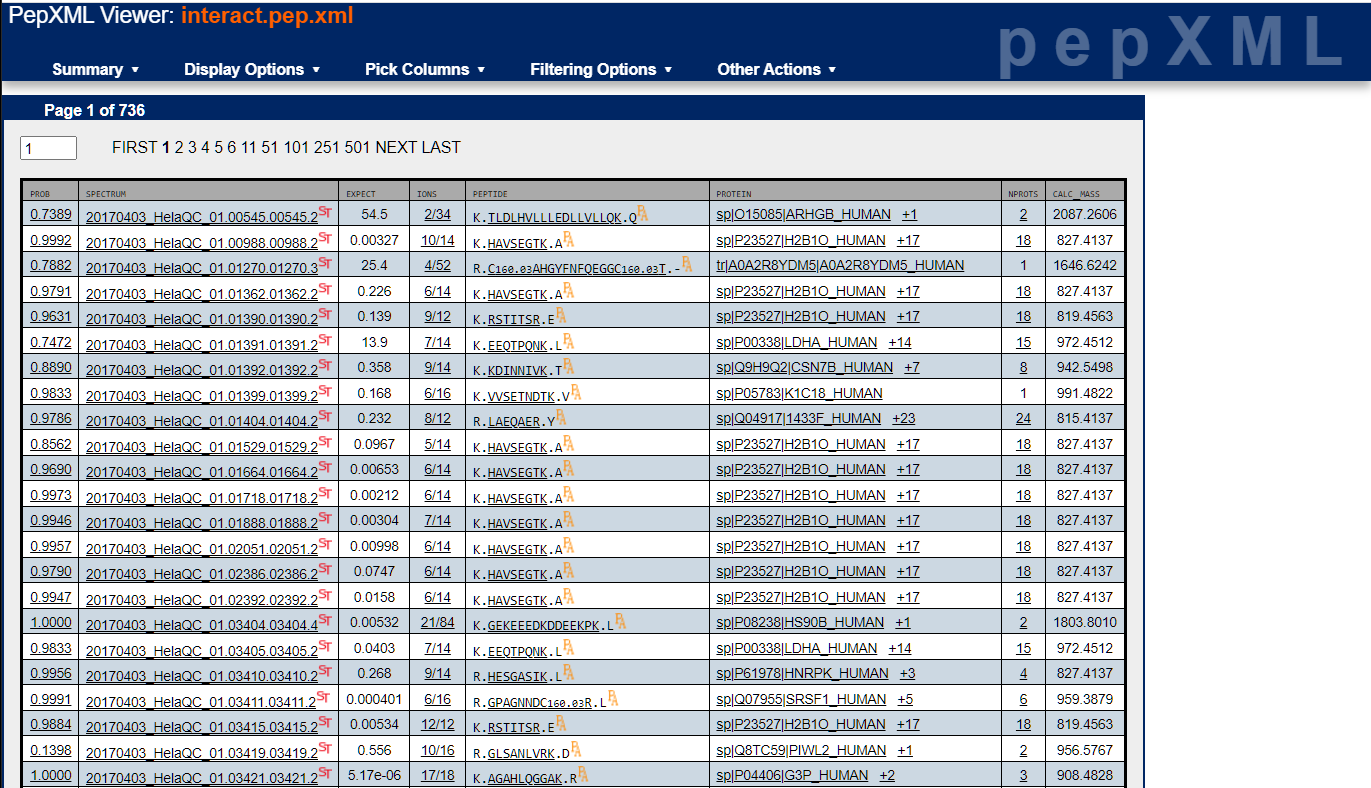
The very first thing I always look at is PeptideProphet's score distribution plots and how well the modeled positive and negative distributions fit the experimental data. To do this, click on the "pep.xml" file link for each analysis. This brings up the TPP's PepXML Viewer below. Think of this as a grid of your raw search results where each row represents an MS/MS spectrum search result. You'll see some scores, a spectrum name with scan number and charge state encoded in it, link to the spectrum viewer, best (not necessarily correct) peptide match, protein name (just one protein name printed but peptide could match to many), and the peptide mass. You can add or remove other columns of information.
Then click on any of the probability score values in the leftmost column with the header "PROB". They will all bring up the same score distribution curves so it doesn't matter which one you click on. You should see a set of model charts like below. There's a lot of information here that is beyond this primer. Your best resource for questions on PeptideProphet and these score distributions is the TPP's support forum.
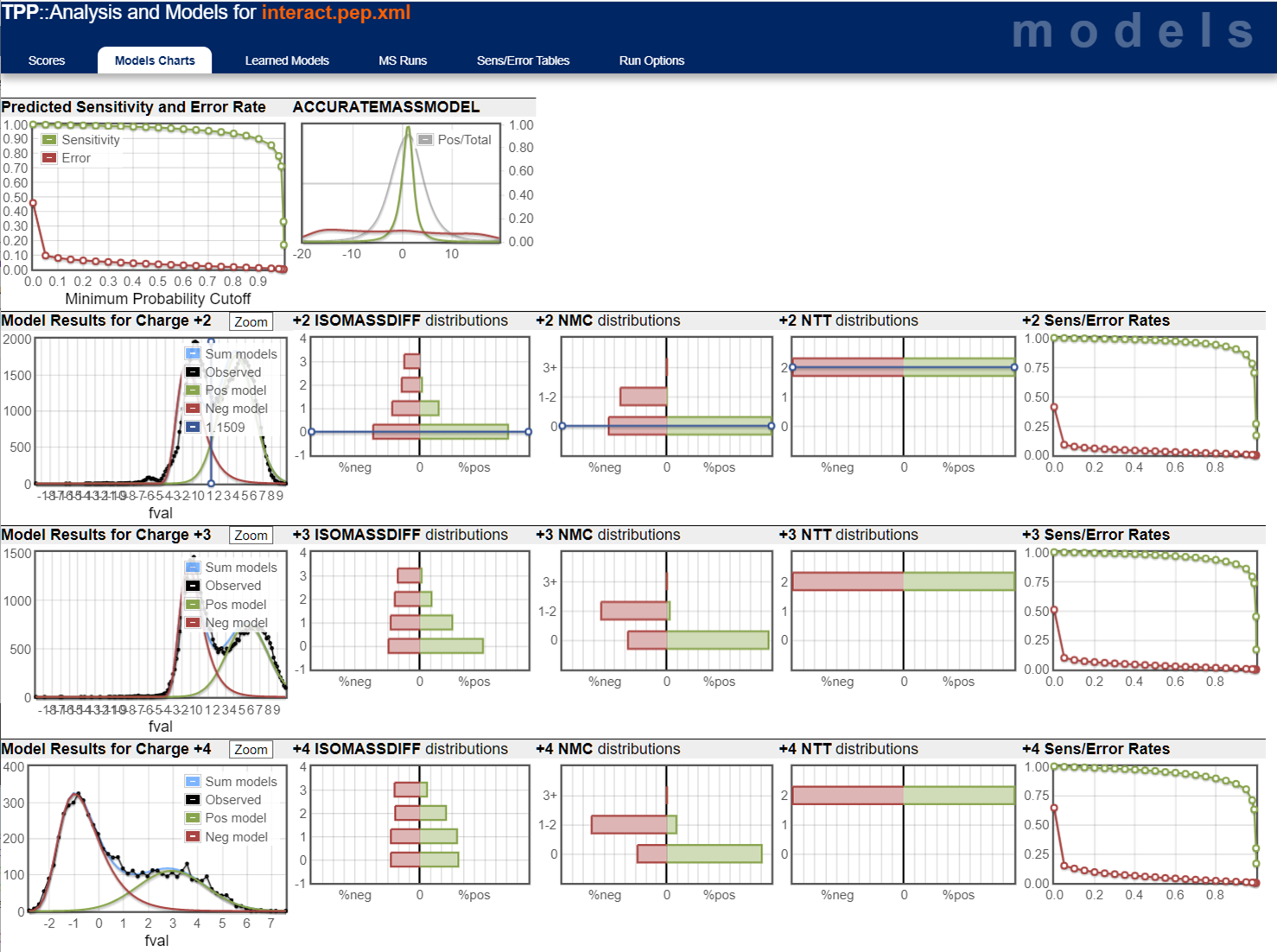
The charts on the far left are the key indicators and what I use to judge whether or not the calculated probability values are worthwhile.
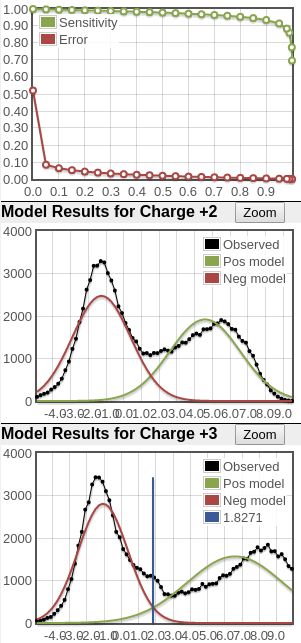
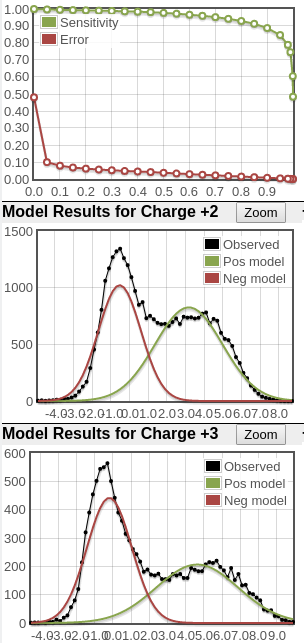
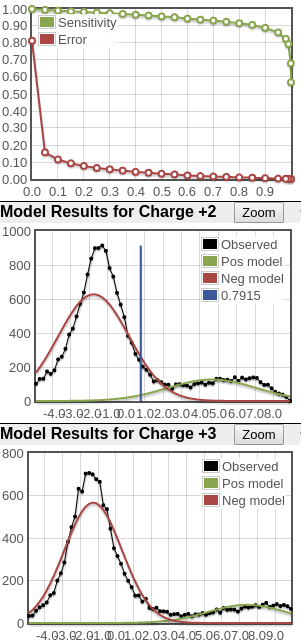
What you see in the plots is a score histogram (black curves) of all the peptide identifications; one plot for each precursor charge state. The red curves are what PeptideProphet fits to the negative/null/wrong distribution and the green curves are what PeptideProphet fits to the positive/correct distribution. What you want to see is that there are two distinct distributions (bimodal) in the black curve and that the red and green curves fit those well. ** NOTE: although each peptide-spectrum-match has a link to the model charts, they all point to the same set of curves; the only difference is the score mark for each specific PSM.
Here's two examples of very good score distributions. Sensitivity/error curves are near ideal (you want them to hit the top right and bottom left corners). The black line in the 2nd and 3rd plots represents histogram of search results and you want to see a bimodal (two peaks) distribution representing the bad hits (modeled by the red curve) and good hits (modeled by the green curve). In these examples, it's clear that there are two peaks in the black score distribution and the positive distributions are big.
Here's two examples of other good, maybe more normal distributions. You can see the positive distributions are not nearly as large as in the plots above but they are clear positive distributions. In the plots on the right, the lines are jagged simply because the raw counts are so low. But even with these low counts, there's good separation between positive and negative distributions (and this good separation is encapsulated in the good sensitivity/error plots).
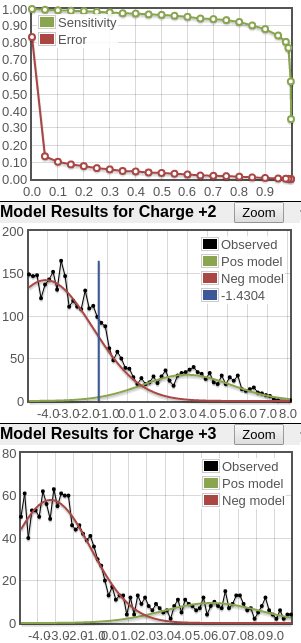
Here's an example of very poor score distributions. The sensitivity/error plots don't trend to the top right and bottom left corners. And there's simply no positive distribution. So if there are any good peptide IDs, their counts are very low.
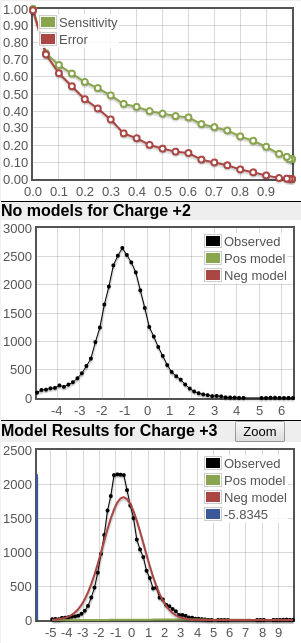
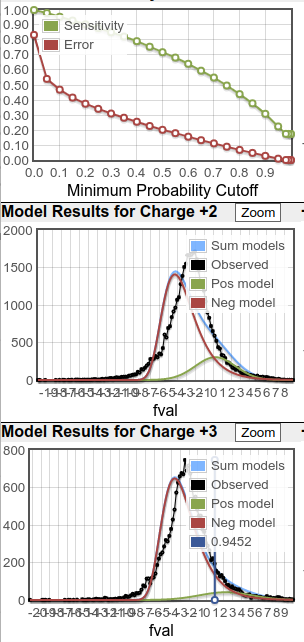
When the score distributions are this poor as in the third example above, I tend to ignore the calculated probability values which means the protein probabilities aren't reliable either (don't bother opening the prot.xml link). For such data, I end up sorting the peptide list in ascending order by the "expect" column (E-value or expectation value) and looking at the best scoring identifications. In contrast to PeptideProphet probabilities calculated by analyzing the entire run, the E-value is calculated on each individual spectrum search; smaller E-values are better. Think of it as related to a p-value but with the definition that it's the expected number of random identifications to score as well or better than the current peptide's score. By observation of Comet scores and the spectral annotations, E-value scores in the range of 10^-8 or smaller are usually very good while spectra for scores in the 10^-4 range and higher start to look more noisy and suspect. And there are always exceptions (like a good looking spectrum match with a poorer E-value). Note that there is an issue where extremely poor matches of say a single fragment ion can generate low E-values so always glance at the "IONS" column as well; any entries with just 1 or 2 fragment matches e.g. "1/20" indicate a poor match.
Both the pep.xml and prot.xml viewers allow you to filter the data. For example, in order to target a specific false discovery rate, you can look up the appropriate probability score cutoff to target that false discovery rate. In the pep.xml viewer, click on any probability score and then click on "Sens/Error Tables". Look at the "Error Table" and the "Error_Rate" column which represents the false discovery rate. If you want to filter the results at a 1% or a 5% false discovery rate, find the "min_prob" minimum probability that corresponds with the 0.01 or 0.05 "Error_Rate" value. Now you can filter your results with this minimum probability cutoff in order to target your specific false discovery rate. (Use the global error rate values and ignore those specific to each charge state.) Click on the images below to get a larger view of the filtering workflows.
pep.xml FDR filtering: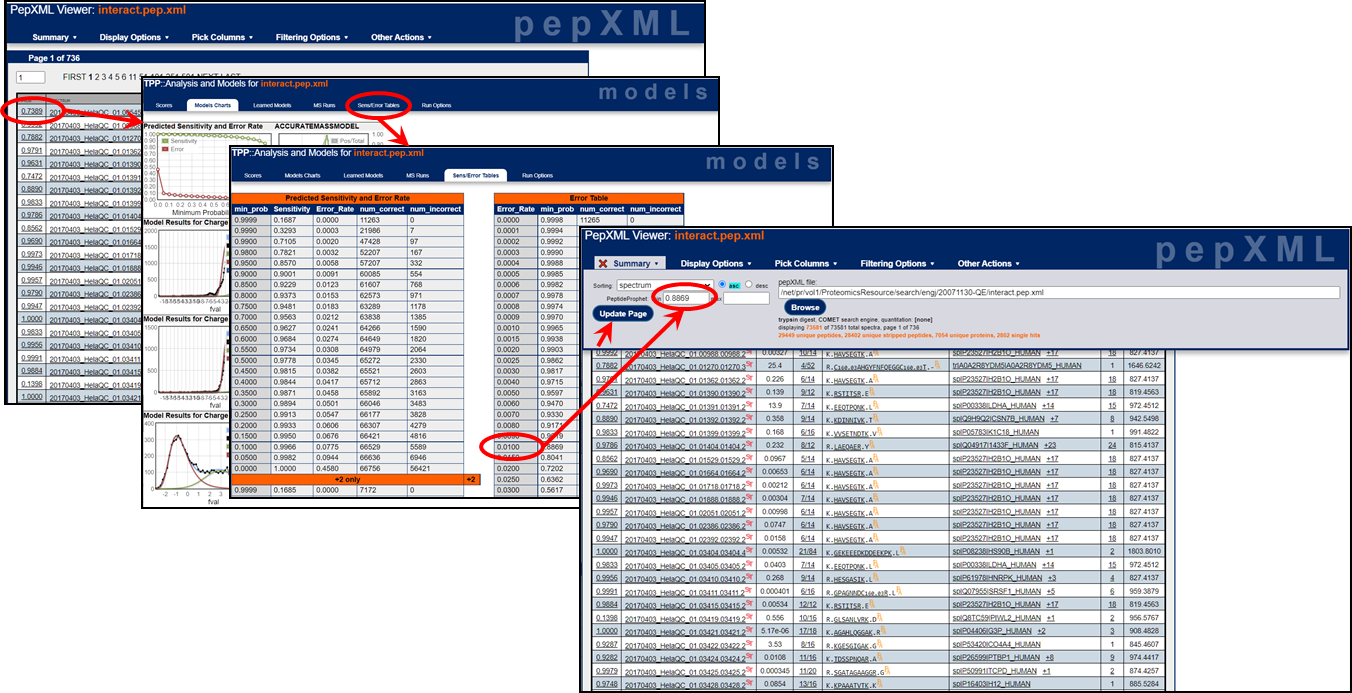
prot.xml FDR filtering: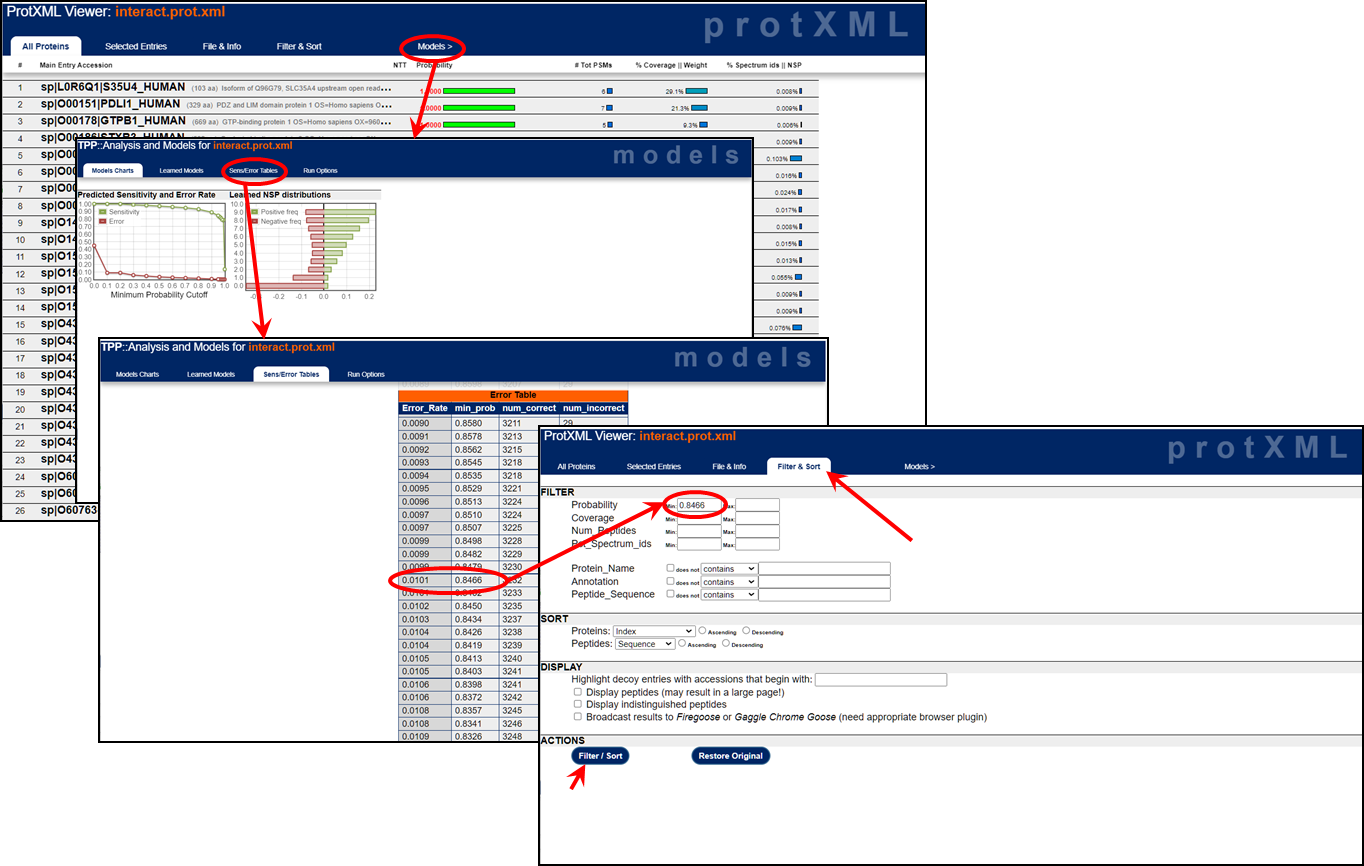
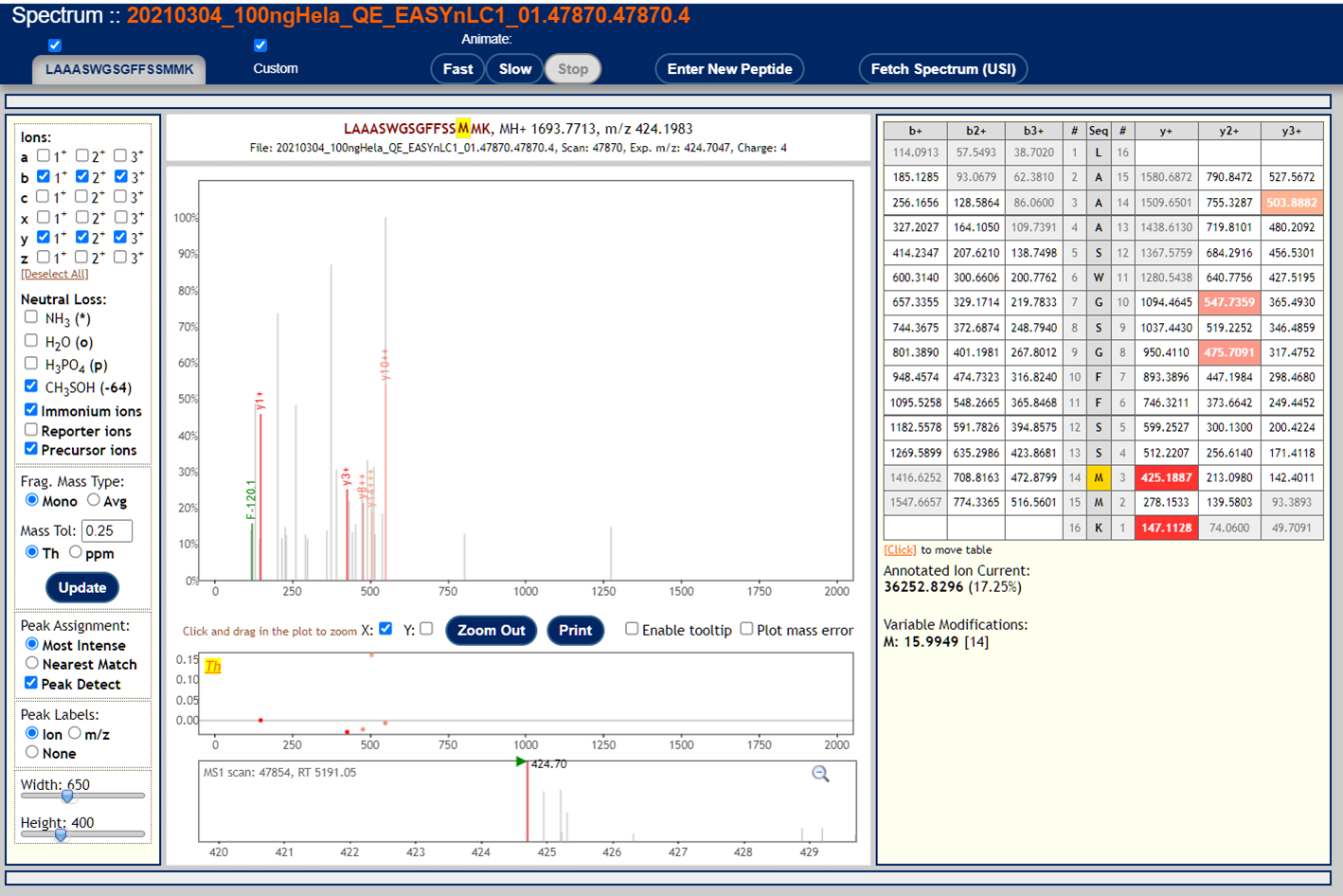
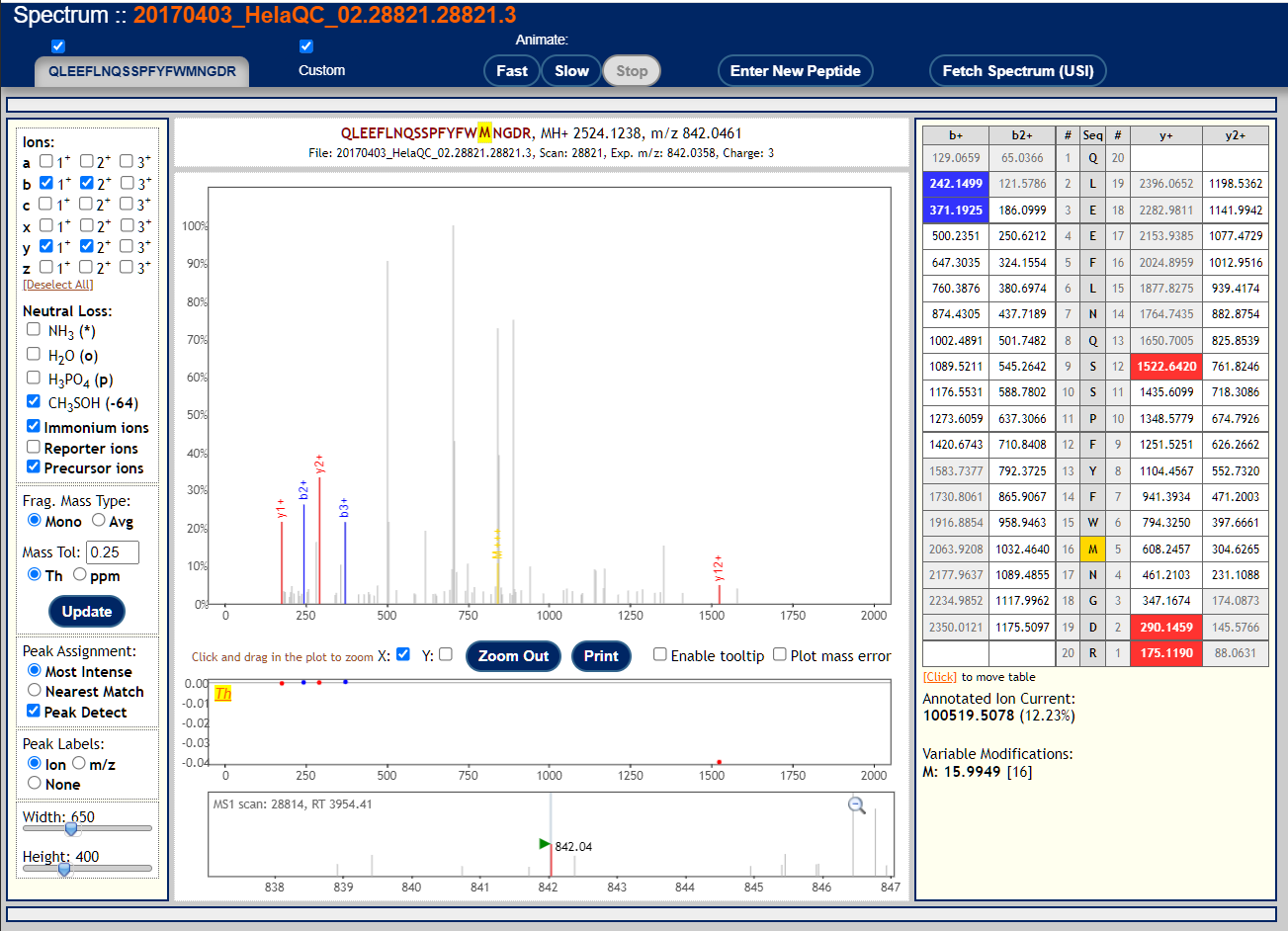
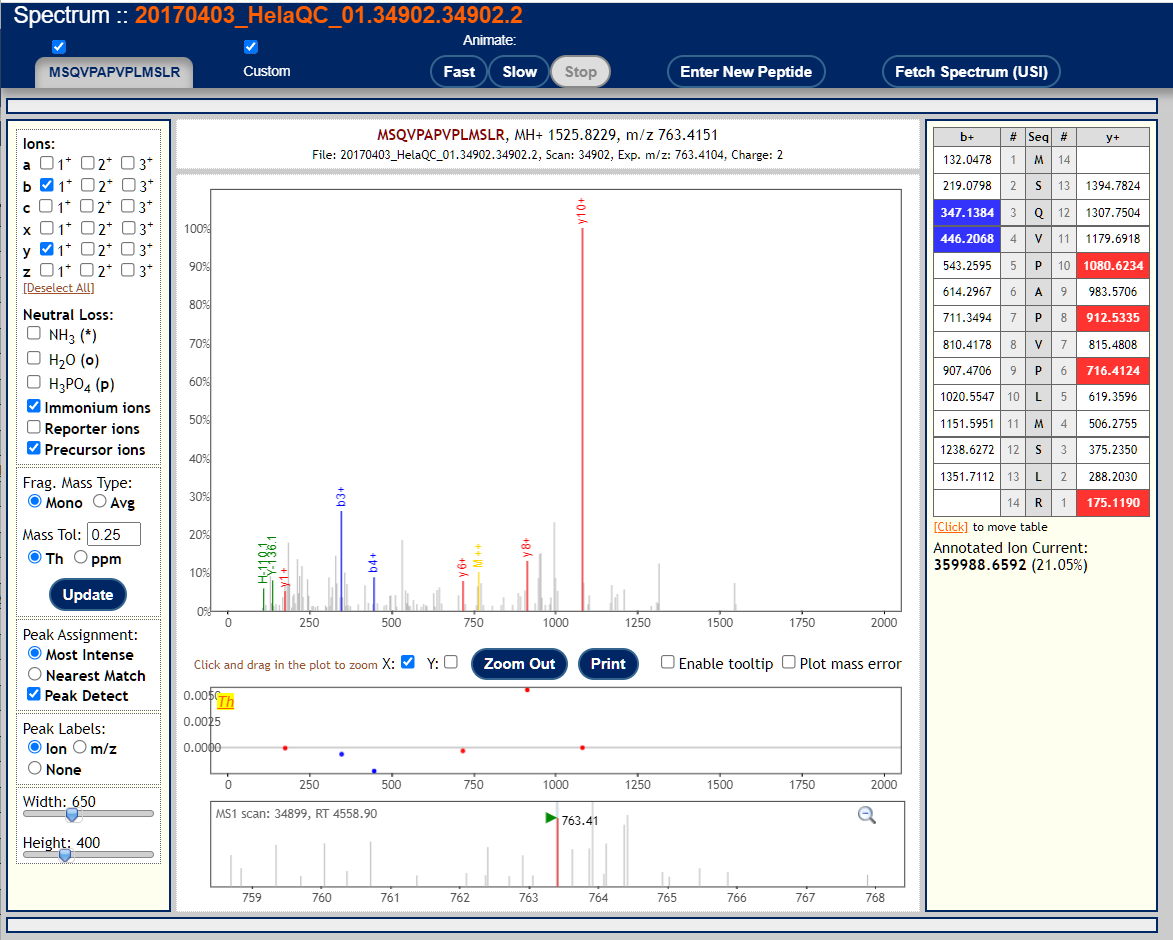
To visualize annotated spectral using the Lorikeet viewer, click on the links in the "IONS" column of the PepXML Viewer. Here are 3 examples of good MS/MS spectra with good peptide matches.
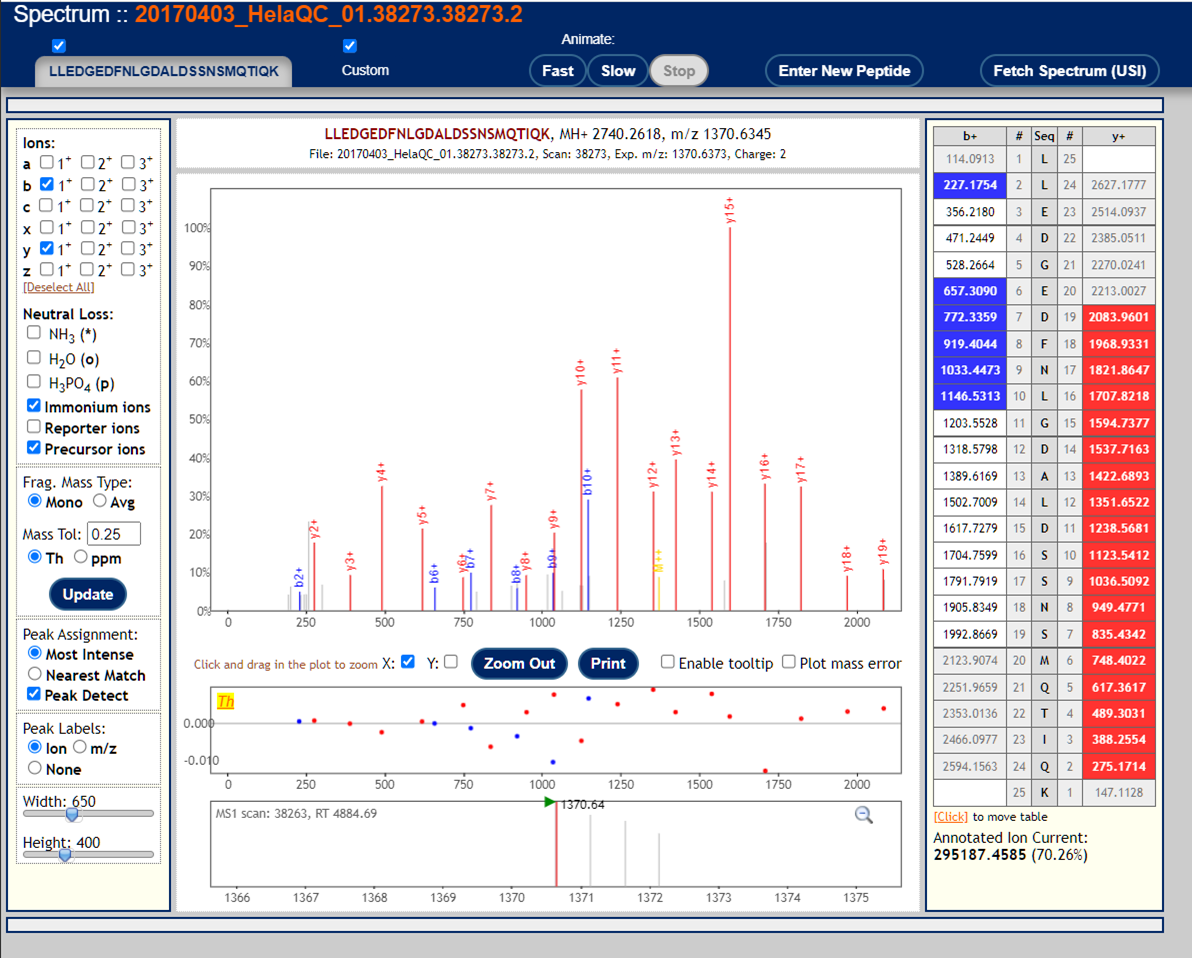
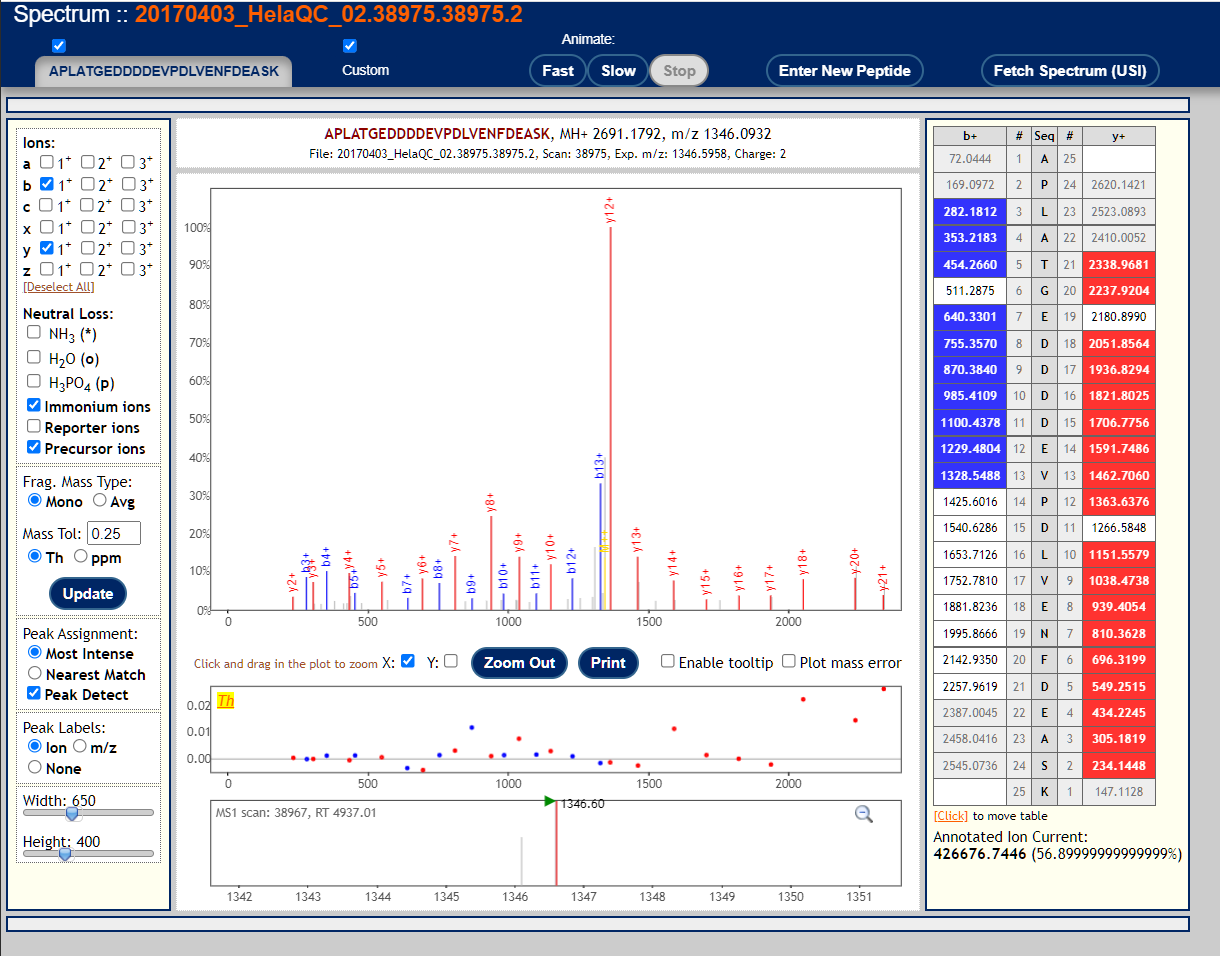
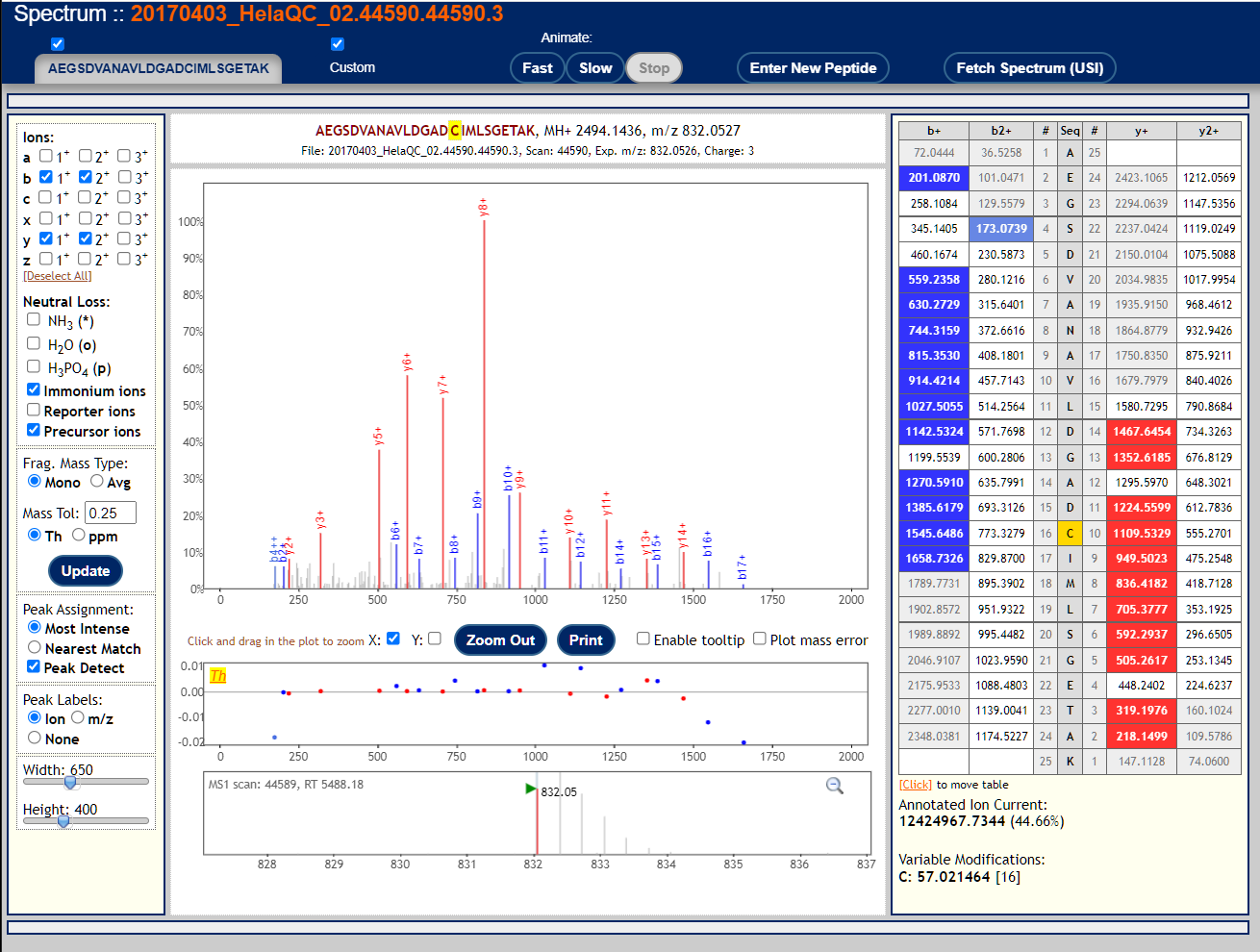
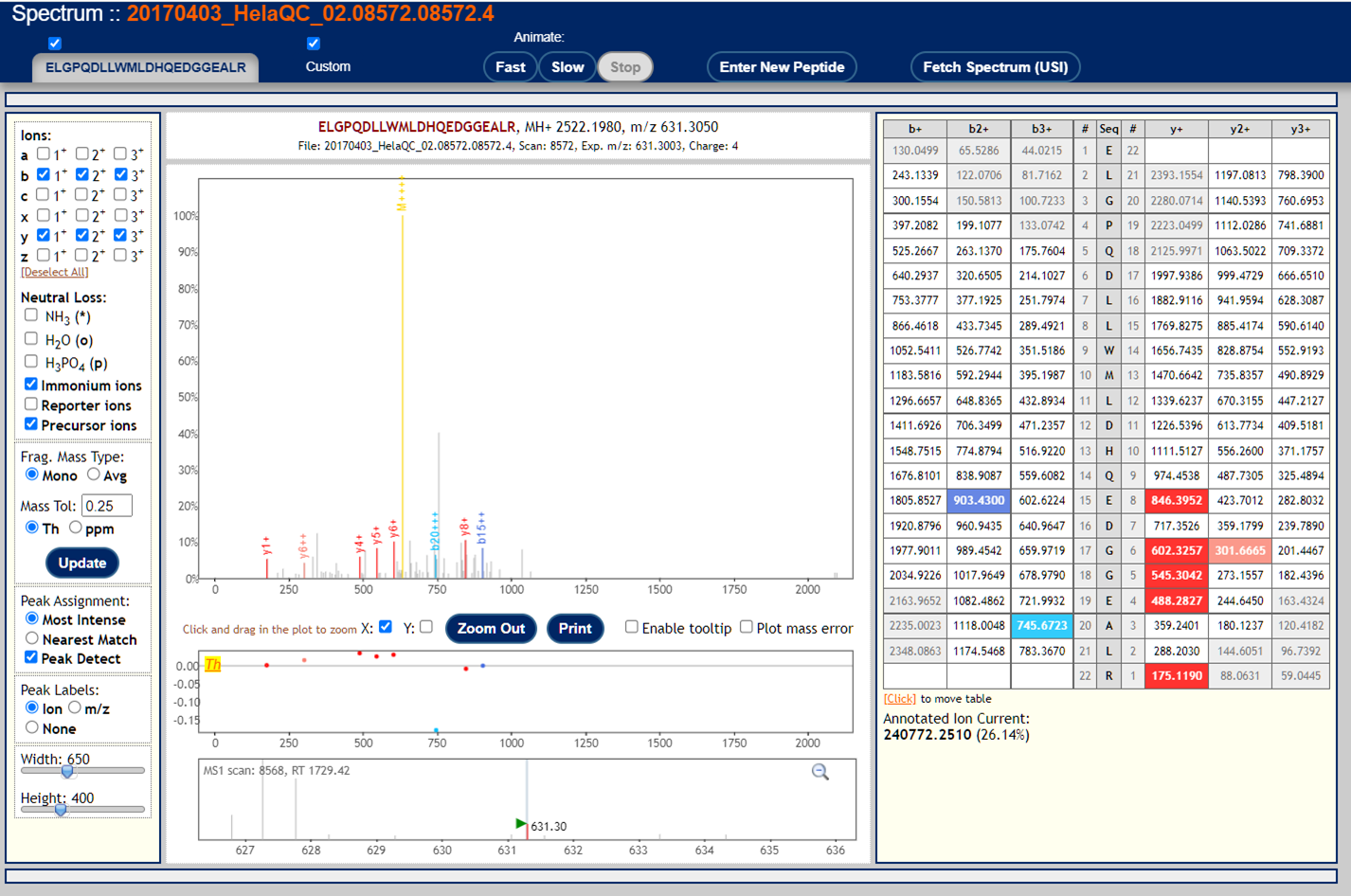
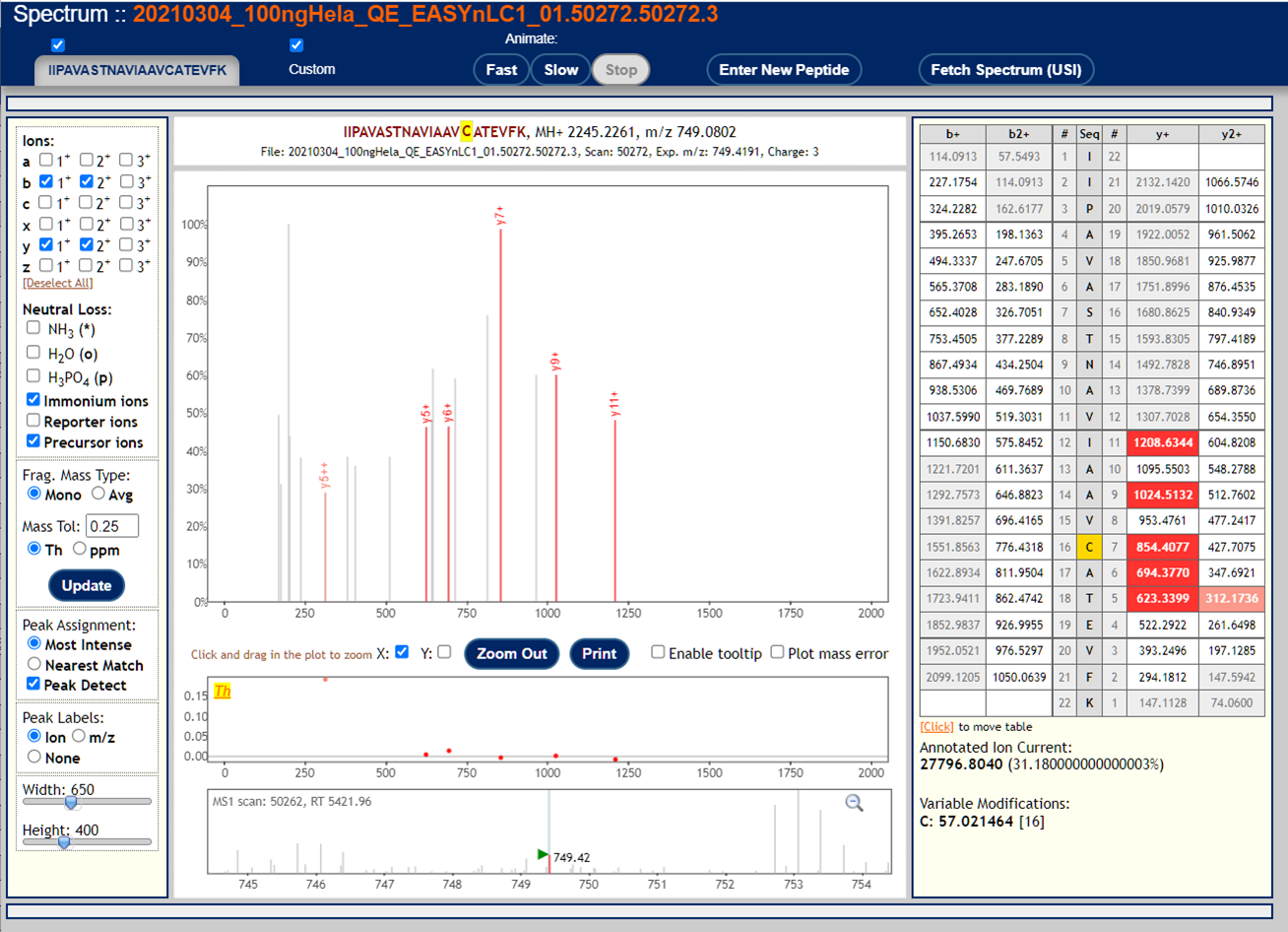
These are examples of very poor MS/MS spectra.
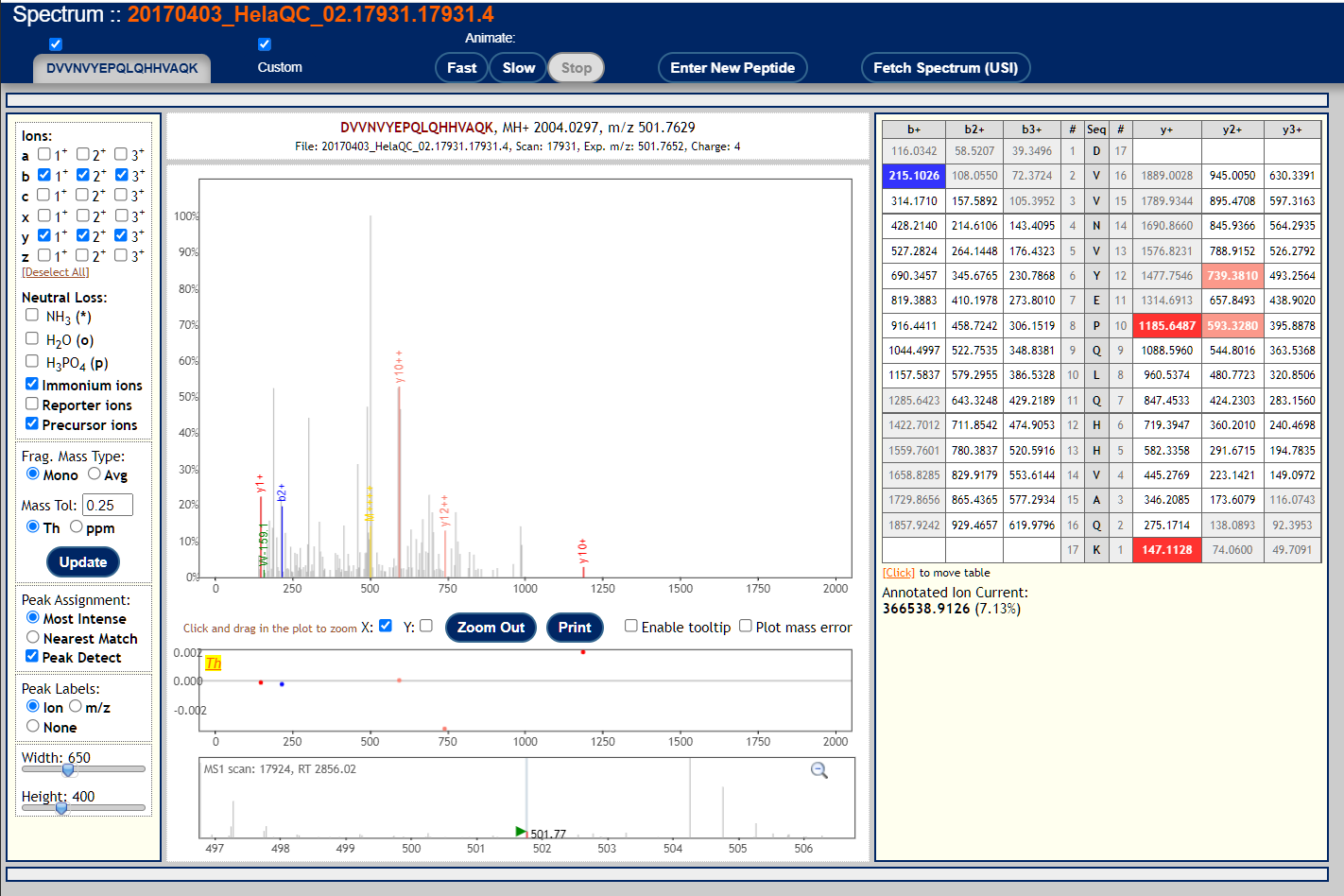
These peptide-spectrum-matches got poor scores but might seem plausible if you squint your eyes hard enough. This reflects the fact that mass spectra are not binary (correct/incorrect) data so identifications and identification confidence are also not black and white.
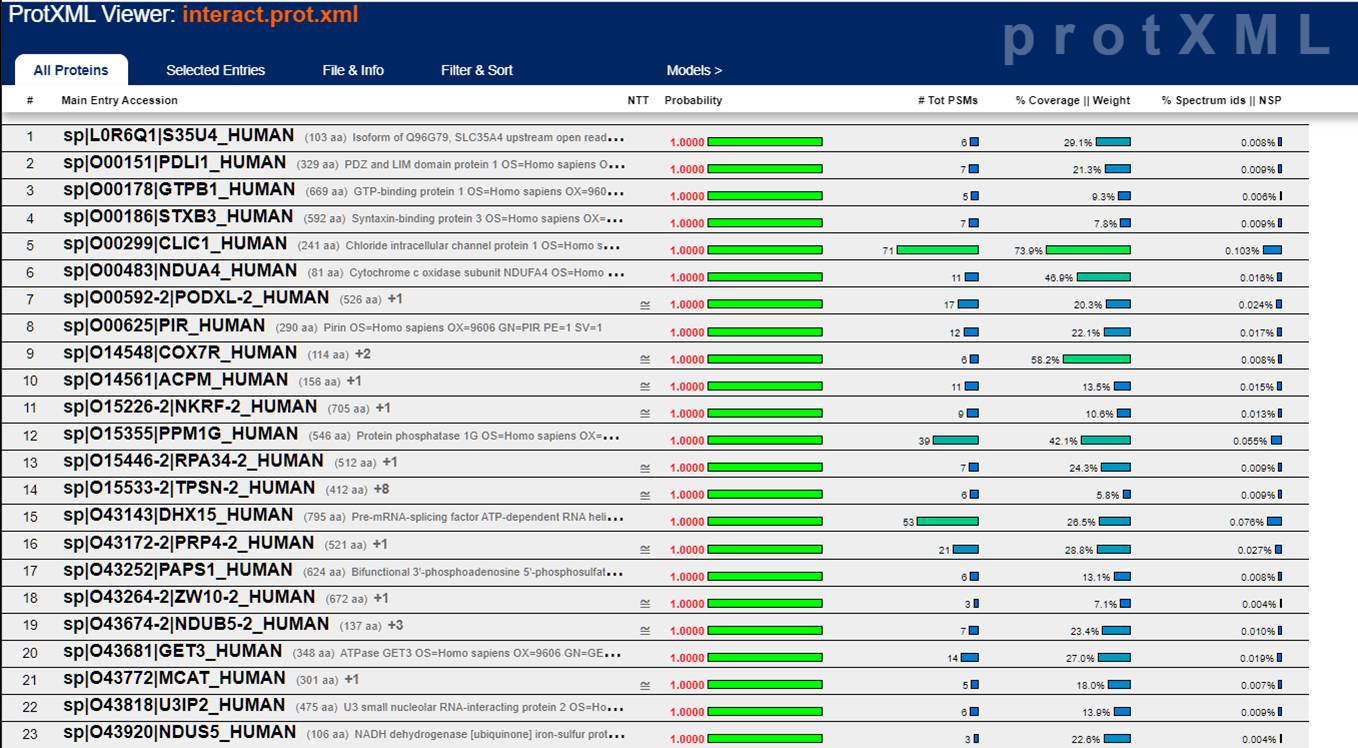
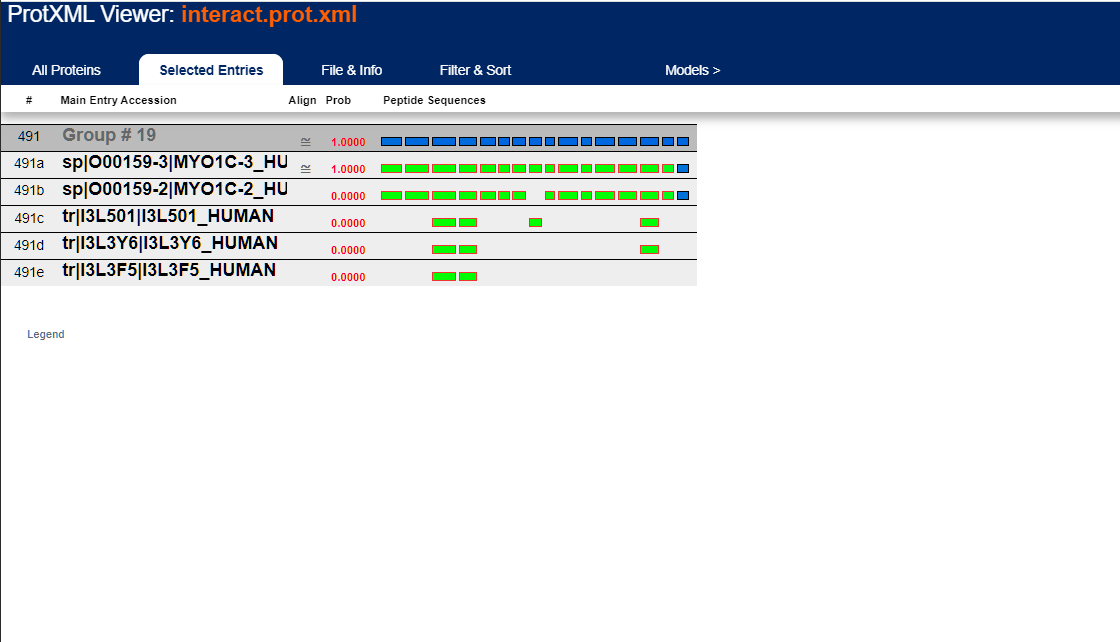
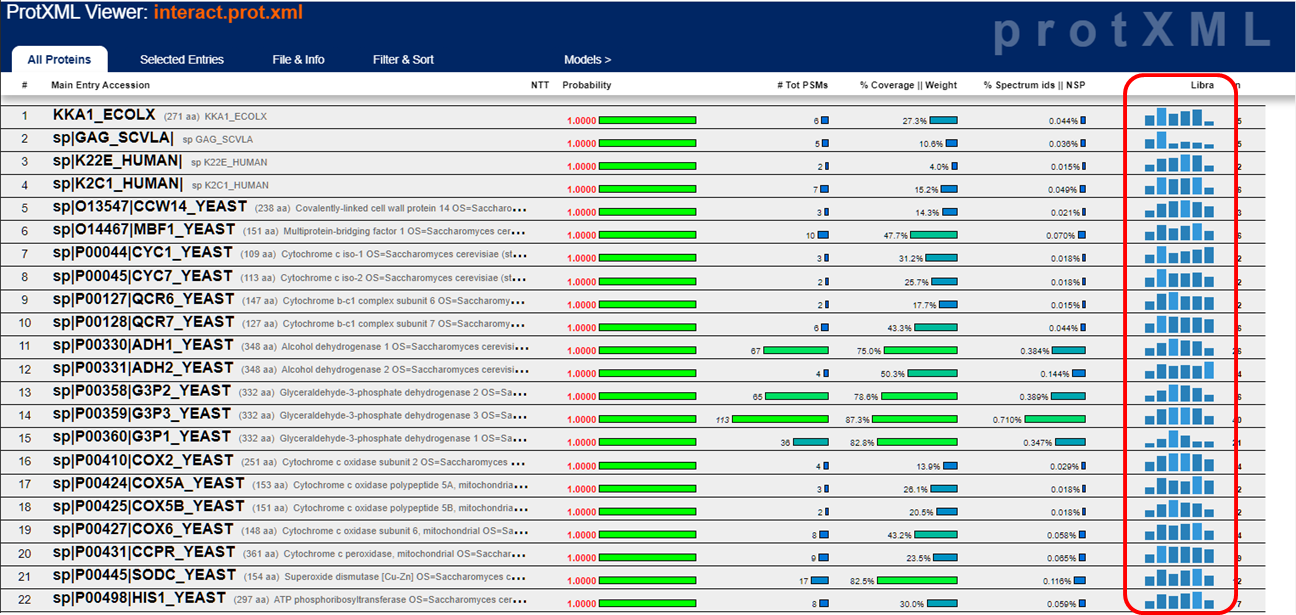
The prot.xml link brings up the ProteinProphet viewer. This is a protein centric view of the data and should only be looked at after you validate that the peptide score distributions aren't horrible. Here's what the ProteinProphet viewer looks like:
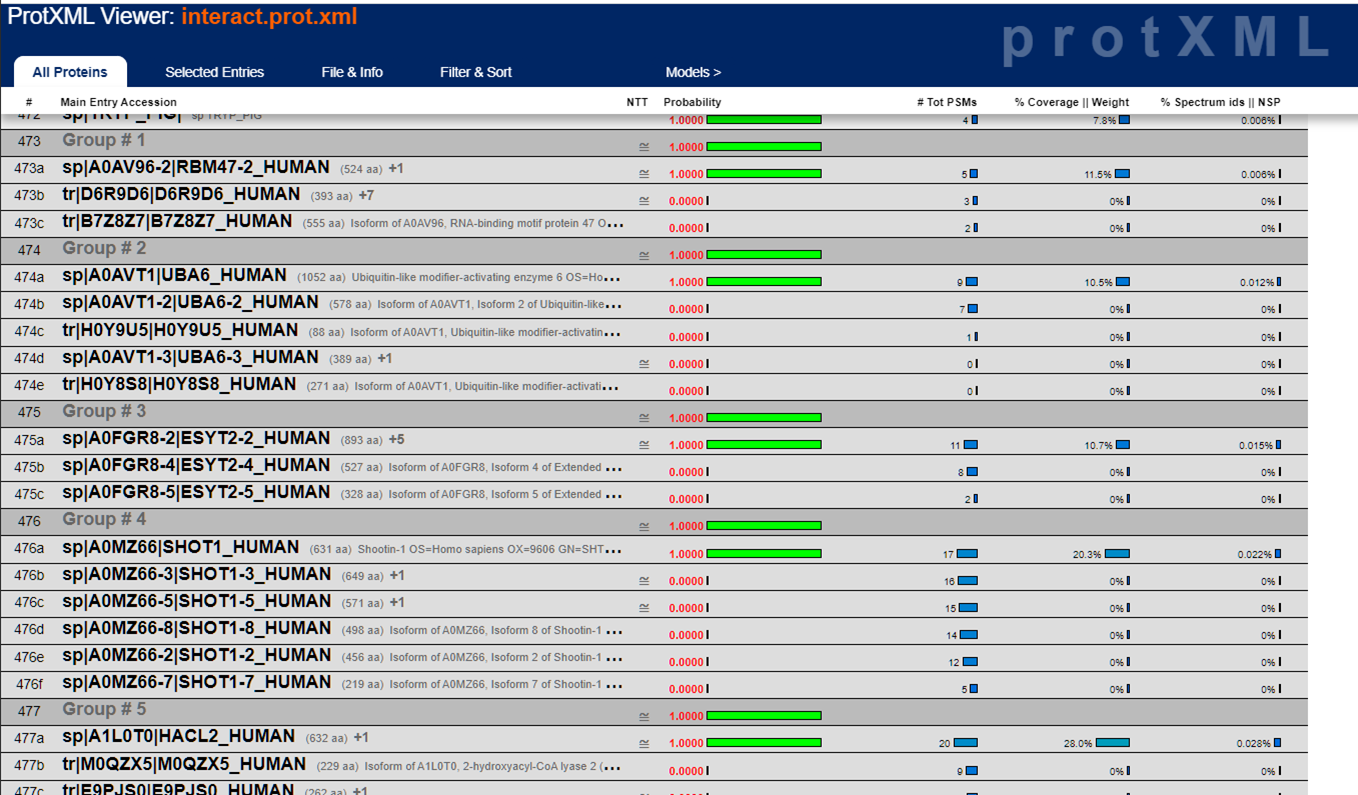
Scrolling down the list, you start seeing protein "groups" which are usually isoforms grouped together (but sometimes they're unrelated proteins that simply share sequence homology):
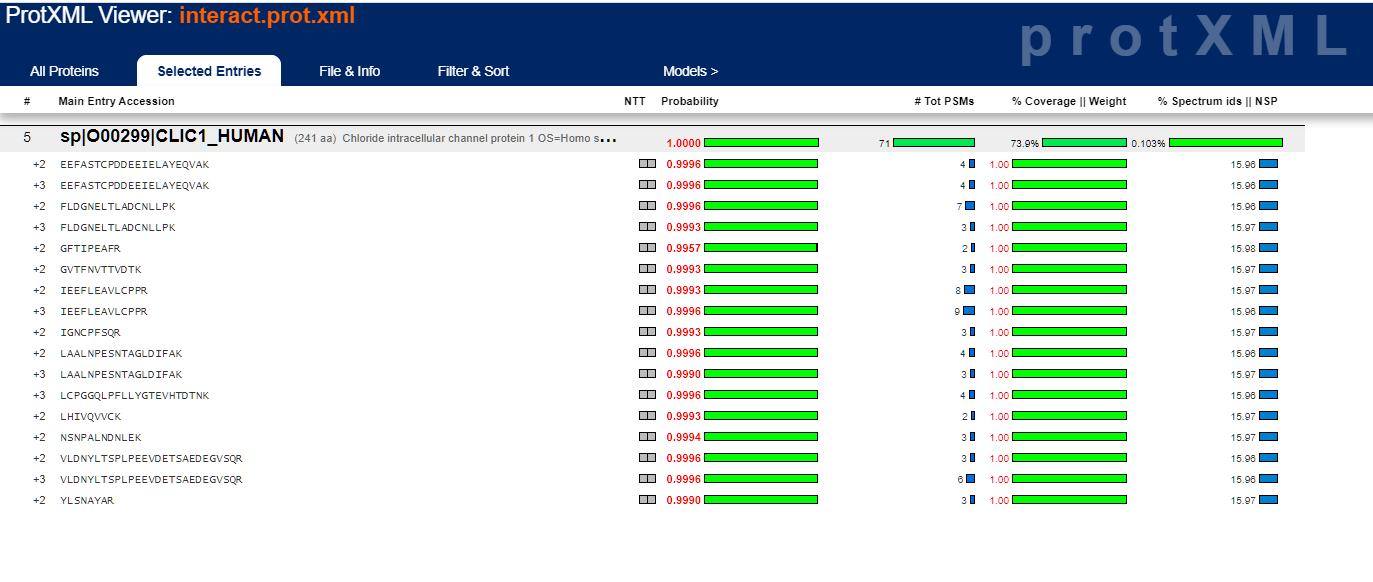
Clicking on the far left column for a particular protein entry will bring up these peptide details:
Clicking on the group entry number will bring up these group details. The shaded boxes represent peptide IDs across the As you can see, entry "sp|O00159-3|MYO1C-3_HUMAN" has contains every peptide identified across this set of 5 proteins where as the other 4 proteins match a subset of those peptides. This is why "sp|O00159-3|MYO1C-3_HUMAN" has a probability of 1.0000 whereas the other 4 proteins have a probability of 0.0000. This doesn't mean that the other proteins aren't present in your sample ... there's no way to know this from the peptide data. The ProteinProphet software will proportion the probability values across the simplest list of protein(s) that can explain the identified peptides which in this case is that is that single protein entry; that simply the convention its authors decided to take. All 5 proteins are still part of the protein group and any or all might be present irrespective of the individual assigned protein probabilities.
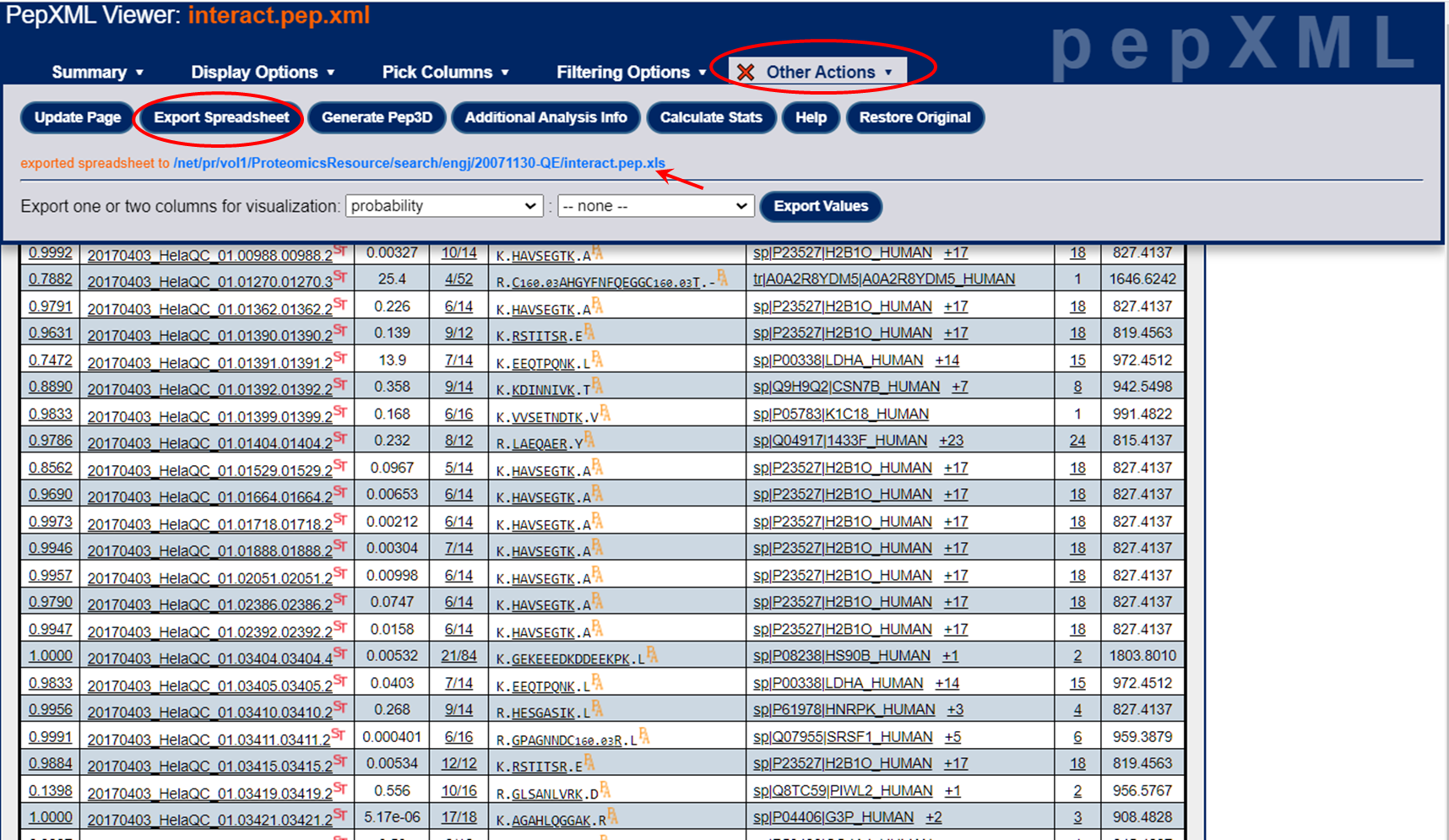
How to download the "Excel" files from the pep.xml and prot.xml viewers:
For the pep.xml link, after you choose "Export Spreadsheet", go back to the "Summary tab". In the header, you should now see an "exported spreadsheet to:" text with a hypertext link to the .xls file (which is really just a tab-delimited text file). See image below. You can just click on the hypertext link to download the .xls file and open in Excel.
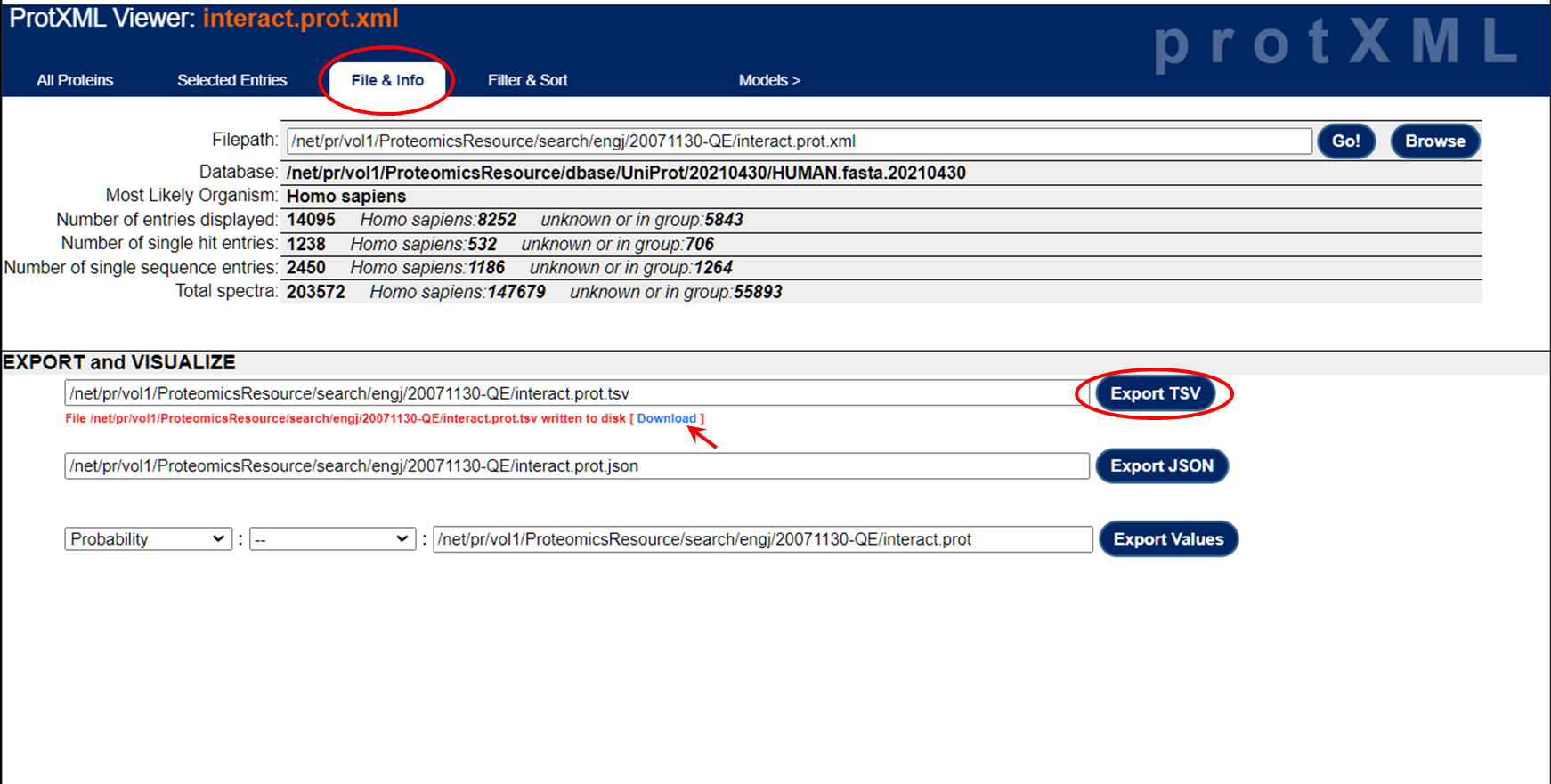
It's a little more convoluted to get the exported file from the prot.xml view. Once you hit the "Export to XLS" button in the protein view, you end up with something like this:
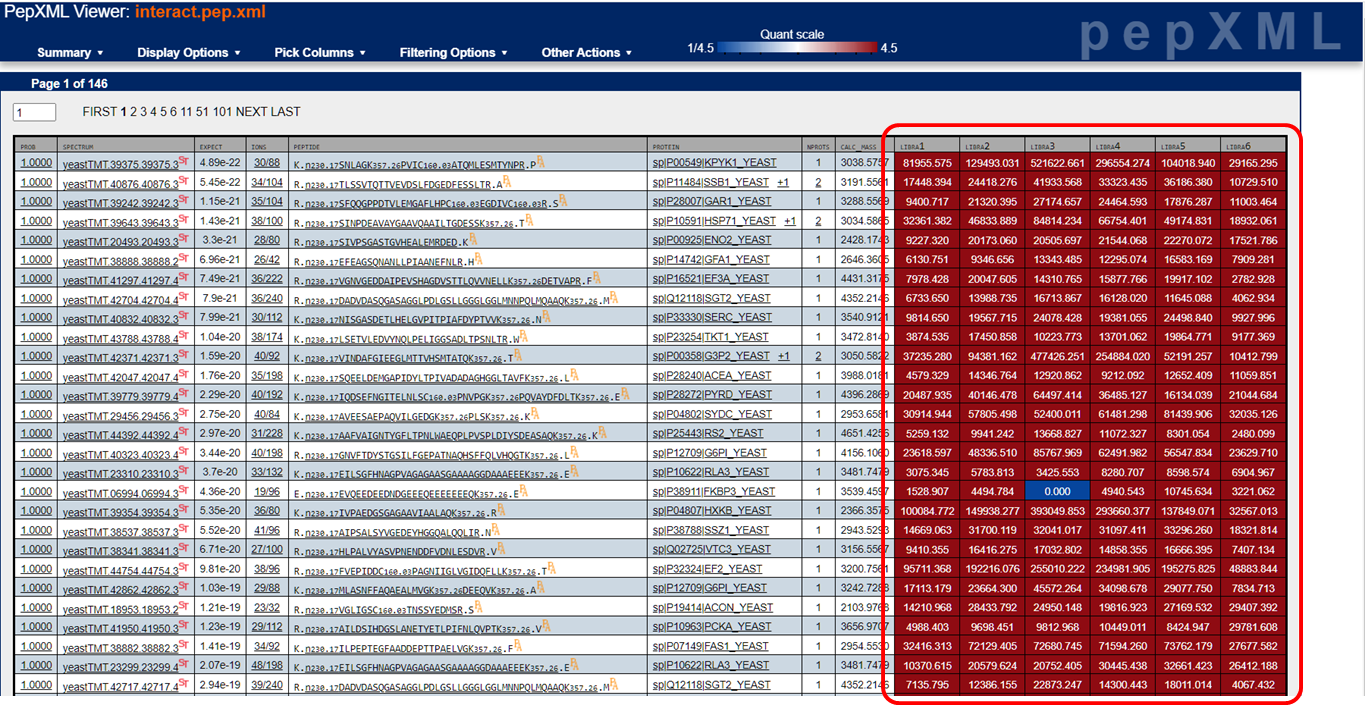
For TMT or iTRAQ labeled samples, the tool that extracts the reporter ions signals is the Libra tool. Assuming the 6-plex TMT reagent was used, the resulting quantitative values would be reported as Libra1 through Libra6 corresponding to reporter ions from 126 through 131. The quantitative peptide and protein values are displayed as below and the actual values will be exported into the tab-delimited text files when exported as described above. Here's the documentation, from back in 2006, that the Libra developers put together describing what it does.
To exclude all 155 contaminants (e.g. trypsin, keratin, albumin, etc.) and decoys from
the PepXMLViewer (pep.xml) viewer, paste this string into the "Required protein text (regex allowed):"
text box of the "Filtering Options" tab. It's one long string, no spaces.